Esta enfermedad se caracteriza por ser un subtipo de gemelos unidos y provocar la duplicación parcial o completa de las estructuras faciales en una única cabeza, cuello, tronco y cuerpo. Esta patología está causada por un defecto de desarrollo durante la embriogénesis poco frecuente y potencialmente mortal, por lo que no tiene tratamiento. Es tan rara que tiene una prevalencia de <1 / 1 000 000
Esta enfermedad, que también recibe el nombre de Síndrome de Kleeblattschadel es una malformación rara en la cual la cabeza presenta un aspecto de trébol de 3 hojas, por lo que los síntomas se pueden apreciar a simple vista. Está provocado por el cierre prematuro de varias suturas, haciéndose ver desde antes del nacimiento. Hasta ahora se han descrito tan sólo 120 casos, con más casos en niñas que en niños. En las formas más graves, el abombamiento bilateral del cráneo en las regiones temporales y en la parte superior provocan un desplazamiento hacia abajo de las orejas, que se disponen de cara a los hombros. Para finalizar, en cuanto al tratamiento se trataría de una técnica neuroquirúrgica para reconstruir correctamente la estructura craneal.
Puedes encontrar más información en el siguiente enlace:
Se trata de una anomalía congénita en la que los conductos intestinal, urinario y reproductor terminan en una cavidad común. Esta enfermedad tiene como causa principal falta de formación del tabique urorrectal durante el desarrollo prenatal.
Este defecto se debe sospechar en un lactante femenino con ano imperforado y genitales de aspecto pequeño. En consecuencia con frecuencia se utiliza una laparatomía, procedimiento para movilizar el intestino y forma de sustitución vaginal durante la reparación.
El gigantismo y la acromegalia
son síndromes caracterizados por la secreción excesiva de hormona de
crecimiento (hipersomatotropismo), casi siempre secundarios a un adenoma
hipofisario.
Antes del cierre de las epífisis, se desarrolla
gigantismo;después de su cierre, acromegalia, que produce características
faciales específicas y otros signos:Los huesos aumentan
de tamaño, incluyendo manos, pies y rostro.
La
hipersecreción de GH suele comenzar entre los 20 y los 40
años. Cuando la hipersecreción de GH comienza después del cierre de las
epífisis, las primeras manifestaciones clínicas son el desarrollo de
características faciales toscas y edema de los tejidos blandos de las manos y
los pies. Entre otros síntomas destacan:
Incrementa el
vello corporal grueso y el espesor de la piel
Sudoración
excesiva debido al crecimiento de las glándulas sebáceas.
El crecimiento excesivo de la mandíbula
produce protrusión del maxilar (prognatismo) y maloclusión de los
dientes.
La proliferación cartilaginosa de la
laringe provoca una voz profunda y ronca. La lengua suele ser grande y
presentar un surco.
La proliferación fibrosa provoca neuropatías periféricas
Las
cefaleas son frecuentes debido a la presencia del tumor hipofisario.
El corazón, el hígado, los riñones, el bazo, la glándula tiroides, las glándulas paratiroides y el páncreas son más grandes de lo normal.
El riesgo de cáncer, en particular del tubo digestivo, aumenta entre 2 y 3 veces.
La GH incrementa la reabsorción tubular de fosfato y produce una hiperfosfatemia leve.
En los pacientes con tumores secretores de GH a menudo se detecta una disminución de la secreción de gonadotropinas.
Ronquidos severos debido a la obstrucción de la vía aérea superior.
En los adultos, los tumores son la causa más común del exceso de esta hormona, los cuales pueden ser pituitarios o no pituitarios.
Cabe destacar que en los pacientes en los que se sospecha acromegalia, debe medirse la
concentración sérica de IGF-1, dado que aumenta típicamente hasta
valores de entre 3 y 10 veces los normales.
Para finalizar, en cuanto al tratamiento de esta enfermedad el método más frecuentes es la
terapia ablativa con cirugía o radiación sobre los tumores hipofisiarios.
Esta enfermedad también conocida tecnicamente como "Epidermolisis Bullosa", se caracteriza por provoca en el afectado una erupción de ampollas en la piel de forma espontánea de contenido seroso o traumatismos mínimos.
Este incremento de la fragilidad de la piel se debe a mutaciones en genes que codifican varias proteínas estructurales, intra o extracelulares, responsables de mantener la resistencia mecánica del tejido en las regiones en donde se encuentran. Las lesiones pueden aparecer en diferentes áreas, siendo las más frecuentes las de las plantas de manos y pies. La evolución de las lesiones se produce en forma de cicatrices y en ocasiones con importantes retracciones. A menudo se acompaña de muchas complicaciones, algunas de las cuales pueden amenazar la vida: importante retraso del desarrollo, gastrointestinales, oftálmicas, laríngeas, dentales, problemas hematológicos, sobreinfecciones generalizadas y deterioro del estado general entre otras. La epidermólisis ampollosa aparece en todos los grupos raciales y étnicos y afecta a ambos sexos por igual. En EE.UU., la frecuencia se estima en 1 de cada 50.000 nacidos. La edad de aparición se sitúa en torno al nacimiento o durante la lactancia. Actualmente, la enfermedad puede diagnosticarse, mediante biopsia de las vellosidades coriónicas de la placenta o la fetoscopia técnica instrumental en la cual se observa directamente el útero para localizar al feto y tomar muestras de sangre y biopsias de piel. En la actualidad no existe un tratamiento curativo, no obstante podemos ayudar siguiendo los siguientes pasos:
evitar los mínimos traumatismos
evitar las sobreinfecciones con cremas con antibiótico a las áreas abiertas
desinfección con clorhexidina al 0,2% y povidona yodada
vendaje de las zonas afectadas y protección con almohadillado de las zonas de presión
vitamina E y corticoterapia en las formas muy graves
reconstrucción plástica funcional
terapia física para minimizar las deformidades.
Puedes encontrar más información acerca de esta enfermedad en los siguientes enlaces:
Esta enfermedad se presenta cuando uno de varios defectos genéticos hace que el cuerpo sea incapaz de producir o distribuir melanina.
Estos defectos se pueden transmitir (ser heredados) de padres a hijos.
Asimismo, existen diferentes formas de albinismo:
Albinismo oculocutáneo: los afectados tienen cabello, piel e iris de color blanco o rosado. También tienen problemas de visión.
Albinismo ocular tipo 1 (OA1): afecta únicamente los ojos. El color de la piel y el de los ojos de la persona generalmente están en el rango normal. Sin embargo, un examen ocular mostrará que no hay pigmento en la retina.
Síndrome de Hemansky-Pudlak (SHP): es una forma de albinismo causada por un cambio en un solo gen. Puede ocurrir con un trastorno hemorrágico, al igual que con enfermedades pulmonares, renales e intestinales
En cuanto a los síntomas, los afectados de esta enfermedad tendrán: falta de color en el cabello, piel o iris del ojo.
En cuanto a los síntomas más característicos encontramos:
Piel y cabello más claros de lo normal
Parches de piel sin color
Ojos bizcos
Sensibilidad a la luz
Movimientos oculares rápidos
Problemas de visión o ceguera funcional
Para diagnosticar dicha enfermedad un médico juzgará la apariencia de la piel, cabello y ojos.
Para finalizar debemos decir que no existe una cura como tal para el albinismo, pues solo podemos proteger ojos y piel del sol.
Puedes encontrar más información acerca de la enfermedad en:
La característica más destacable de esta enfermedad es el crecimiento de pelo, tanto en cantidad como en grosor, en zonas del cuerpo donde habitualmente no crece, siendo un problema estético que no supone ningún riesgo para la salud. Cabe destacar que las zonas donde crece más pelo es en toda la superficie corporal, salvo las plantas de los pies y las palmas de las manos.
Los principales mecanismos causantes de la hipertricosis son tres: la conversión de vello en pelo terminal, cambios en el ciclo del crecimiento del pelo o incremento de la densidad de los folículos pilosos.
No obstante, aunque estamos hablando de una enfermedad rara, distinguimos diferentes tipos:
Lanuginosa: cabello liso y sedoso que crece en todo el cuerpo de manera uniforme. se adquiere genéticamente.
Congénita: es la conocida como "síndrome de hombre lobo" y está ligada al cromosoma x. Este cabello es más fuerte y aparece en la cara.
Lumbosacral: recibe el nombre de cola de fauno, ya que el pelo crece exclusivamente en la zona lumbosacral.
Iritativa: se adquiere como consecuencia de un trabajo frecuente y repetitivo al que se somete una parte determinada del cuerpo como pasar mucho tiempo de rodillas.
Nevus: la hipertricosis puede estar asociada a la presencia de un nevus, en cuyo caso suele ser indicativo de que probablemente éste no se transformará en un melanoma.
El único tratamiento posible para la hipertricosis es el afeitado o la depilación.
Se trata de una enfermedad muy rara, pudiendo suceder una vez de cada 50000 nacimientos. Esta enfermedad se caracteriza por la formación de un ser humanoide hospedado en alguna zona del feto que sí ha nacido y que al principio se muestra como un bulto en a zona donde se situará el parásito. El lugar más común en que se hospeda el feto es el abdomen, aunque también pueden encontrarse en : escroto, hígado, riñones y en los casos más graves en la cavidad craneal.
Generalmente estos casos se diagnostican los primeros años de vida o incluso durante el embarazo mediante ecografías, pero como son muy raros también pueden pasar desapercibidos hasta que el feto atrofiado crece.
Casi siempre esta enfermedad se diagnostica como una tumoración no dolorosa llamada teratoma,compuesta de tejidos poco organizados que derivan de las tres capas embrionarias, aunque también se han encontrado diferenciados .
En la actualidad la mayoría de los autores consideran que fetus in fetus y teratoma no son distintas entidades, si no dos aspectos de la misma patología en diferentes estados de maduración.
La causa de esta enfermedad es un error genético durante la fecundación del óvulo; esto origina que la segmentación de las células no sea correcta y que los dos gemelos no lleguen a separarse completamente, si no que quedan unidos por alguna zona del cuerpo. Uno de ellos crece sano y sin problemas, mientras que el otro se atrofia en el interior de su hermano y depende completamente de él, por lo que se le llama “gemelo parásito”
De esta enfermedad apenas se conocen unos 100 casos en el mundo.
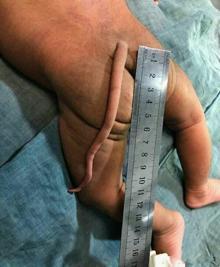
Los afectados presentan en la zona final del sacro,a la altura de cóccix, una "cola". Cabe destacar que no es exacta a la presente en otros animales mamíferos ya que esta contiene hueso, cosa que en los afectados no ocurre. La cola vestigial en humanos va a estar compuesta de tejido conectivo, músculos, vasos sanguíneos, nervios y piel (sólo en algunos casos estaría compuesto por cartílago y vértebras).
Normalmente al nacer se extirpa, pero a veces algunos adultos la conservan.
La causa de esta cola vestigial es una mutación genética que reactiva un carácter oculto de nuestro desarrollo evolutivo. Gracias al descubrimiento de los genes que controlaban el desarrollo en otros vertebrados, se pudo comprobar que estos se encontraban también en el genoma humano, no obstante es por la regulación y apoptosis de las células que estaban destinadas a formar una cola, por lo que a pesar de contener esos genes, no se llega a desarrollar en la mayoría de los casos.
Esta enfermedad congénita causa un crecimiento excesivo de la piel y un desarrollo anormal de huesos, músculos, tejido adiposo, vasos sanguíneos y linfáticos, normalmente acompañados de tumores en más de la mitad del cuerpo. No obstante esta enfermedad es tan rara que sólo se han confirmado poco más de 200 casos en todo el mundo.
El origen de la enfermedad es inconcreto Hasta el momento no se le ha podido encontrar el origen directo a este síndrome, pero al parecer todo indica que la condición radica en una mutación del gen supresor tumoral PTEN .
Es una enfermedad progresiva ya que los niños suelen nacer sin ninguna deformidad evidente. Conforme crecen aparecen los tumores y el crecimiento de la piel y de los huesos. La gravedad y la localización de estos crecimientos asimétricos varían ampliamente, aunque suelen darse en el cráneo, uno o más miembros y en la plantas de los pies.
Además, existe un riesgo de muerte prematura en los individuos afectados debido a trombosis y tromboembolismo pulmonar, causadas por malformaciones asociadas a este desorden en los vasos.
El síndrome de Fanconi es una patología en donde substancias que deberían ser absorbidas de manera normal por los túbulos renales no lo son ya que estos no son capaces de hacerlo y en su defecto son expulsadas por la orina. Los principales compuestos son glucógeno, fructosa, galactosa, cistina (que es un aminoácido de la queratina; esto produce cistinosis, enfermedad grave caracterizada por acumulación de esta sustancia en lisosomas, lo que provoca daño en distintos órganos y tejidos como ojos).
Si esta patología aparece en niños suele deberse a trastornos genéticos, sin embargo en avanzadas edades suele ser por otros problemas.
Como síntoma muy característico suele ser una sed exacerbada y una continua diuresis.
El síndrome de Fanconi no tiene curación pero se puede controlar para mejorar la calidad de vida. La principal indicación es beber bicarbonato sódico para contrarrestar la acidosis sanguíneo asi como la ingesta de potasio en caso de que su valor este bajo.
En los casos más graves, el transplante de riñón puede ser una opción muy favorable.
Esta enfermedad también conocida como "progeria" es un trastorno genético progresivo que acelera el envejecimiento de los niños y que comienza en los primeros dos años de vida.
Esta patología se debe una mutación en el gen LMNA, la cual participa en el soporte estructural del núcleo celular, conllevando el envejecimieno prematuro.
Es durante el primer año cuando comienzan a aparecer los signos y síntomas, como crecimiento lento y caída del cabello.No obstante, las primeras causas en la muerte de los niños que padecen esta enfermedad son los problemas cardíacos o los accidentes cerebrovasculares siendo la esperanza de vida de 13 años aproximadamente.
La progeria no tiene cura, pero las investigaciones en curso muestran cierta esperanza en cuanto al tratamiento.
Se trata de una “genodermatosis”rara hereditaria caracterizada por una infección crónica por el virus del papiloma humano (VPH), que conlleva la aparición de lesiones cutáneas polimorfas y un riesgo elevado de cáncer de piel.
La EV se debe a mutaciones de pérdida de función de uno de los dos genes EVER1/TMC6 y EVER2/TMC8 (17q25.3).
Se inicia durante la niñez, como una dermatosis diseminada con predilección por áreas expuestas, como cara (frente), cuello, dorso de las manos y los dedos, caras externas de antebrazos, tronco y extremidades inferiores. Después se presenta diseminación, bilateral y simétrica. Está constituida por muchas neoformaciones de 2 a 5 mm de diámetro, redondeadas, ovales o poligonales, aisladas, del color de la piel, con superficie lisa o cubiertas de escamas finas.
El diagnóstico se basa en los hallazgos clínicos e histológicos. Una biopsia cutánea evidencia lesiones similares a verrugas planas con leve hiperqueratosis, hipergranulosis y acantosis de la epidermis.
El tratamiento de elección para el CCE es la escisión quirúrgica. La prevención es crucial en el manejo de la enfermedad (evitar la exposición al sol, fotoprotección). El pronóstico es favorable ya que el cáncer de piel evoluciona lentamente y las metástasis son raras.